 - Mdr Cert")
What Is MDR (Medical Device Regulation)?
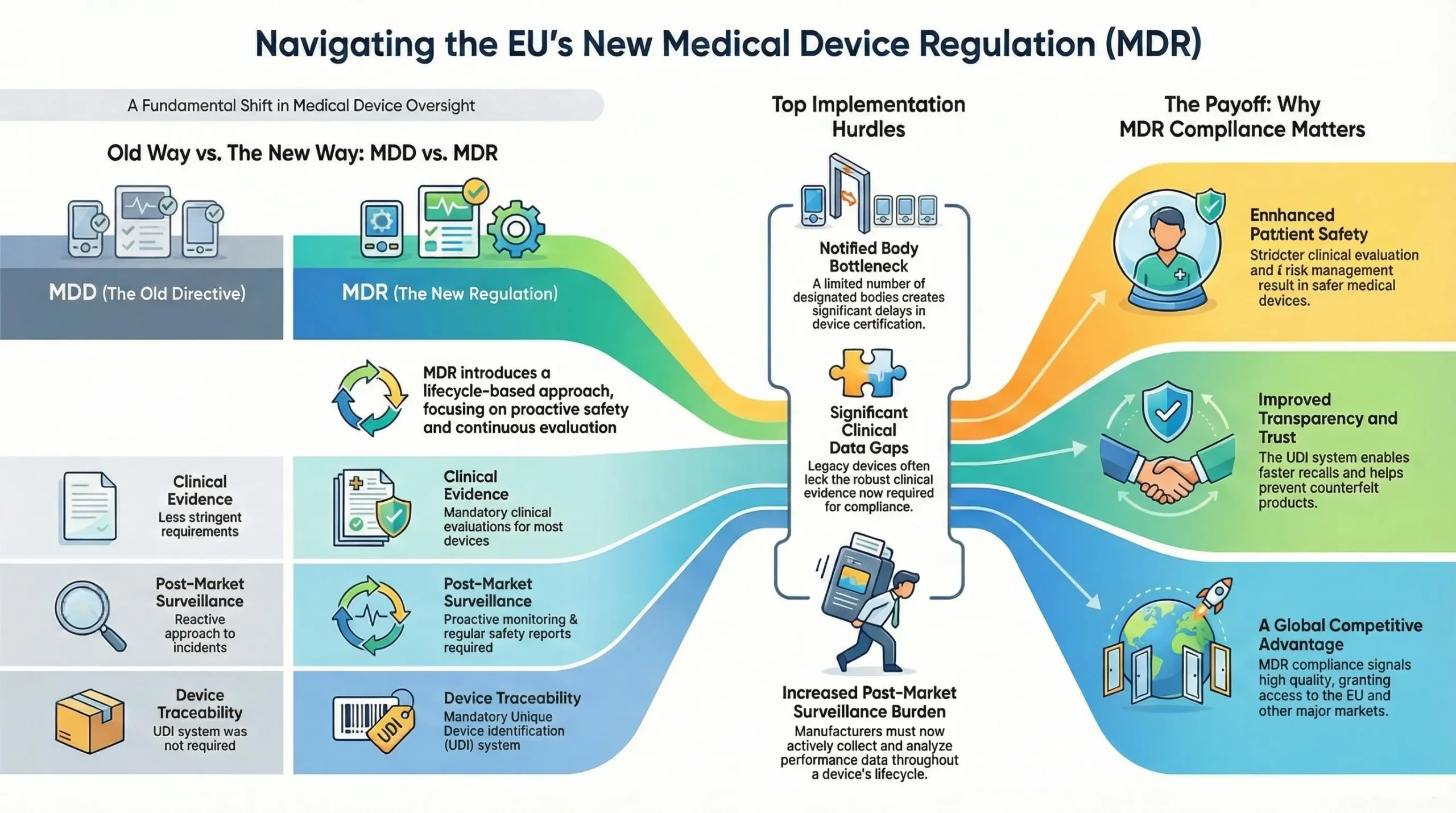
The Medical Device Regulation (MDR), officially known as EU Medical Device Regulation (MDR 2017/745), is a regulatory framework established by the European Union (EU) to oversee the safety, performance, and quality of medical devices in the European market. Replacing on May 26, 2021 the previous Medical Devices Directive (MDD 93/42/EEC) , the MDR introduces stricter requirements for manufacturers, notified bodies, and distributors to ensure better patient safety and transparency.
The MDR applies to a wide range of medical devices, from simple bandages to advanced implants and diagnostic software. Its primary goal is to enhance post-market surveillance, improve clinical evidence requirements, and strengthen traceability through the Unique Device Identification (UDI) system.
Key aspects of MDR include:
- Stricter clinical evidence requirements
- Enhanced post-market monitoring
- Unique Device Identification (UDI) system for traceability
- More rigorous notified body oversight
Why MDR Matters for Medical Device Manufacturers
The MDR (2017/745) significantly impacts medical device manufacturers by enforcing more rigorous compliance standards. Here’s why it’s essential:
- Enhanced Patient Safety – The MDR imposes stricter clinical evaluation and post-market surveillance to minimize risks associated with medical devices.
- Greater Transparency – Manufacturers must provide detailed technical documentation, making device information more accessible to healthcare professionals and patients.
- Stricter Certification Process – Notified bodies face more stringent assessments, ensuring only compliant devices enter the EU market.
- Improved Traceability – The UDI system helps track devices throughout their lifecycle, enabling faster recalls if safety issues arise.
- Global Market Alignment – Complying with MDR helps manufacturers meet international regulatory expectations, facilitating market expansion.
For manufacturers, non-compliance means losing access to the EU market, one of the largest medical device markets globally. Staying ahead of MDR requirements is not just a legal obligation—it’s a competitive advantage.
History of Medical Device Legislation in the EU
The EU’s medical device regulations have evolved from:
- Active Implantable Medical Devices Directive (AIMDD, 1990)
- Medical Devices Directive (MDD, 1993)
- In Vitro Diagnostic Devices Directive (IVDD, 1998)
The MDR (2017/745) fully applicable since May 2021, was introduced to address gaps in the MDD, particularly concerning high-risk devices and post-market surveillance.
Key Differences Between MDD and MDR
| Aspect | MDD (93/42/EEC) | MDR (2017/745) |
| Scope | Limited software regulation | Explicit inclusion of software as a medical device |
| Clinical Evidence | Less stringent requirements | Mandatory clinical evaluations for most device |
| Post-Market Surveillance (PMS) | Reactive approach | Proactive PMS & Periodic Safety Update Reports (PSURs) |
| UDI System | Not required | Mandatory for traceability |
| Notified Body Oversight | Less rigorous | Stricter assessments & unannounced audits |
Which Products Fall Under MDR?
Under the EU Medical Device Regulation (MDR 2017/745), medical devices are classified into four main risk categories. This classification is based on factors such as the invasiveness of the device, duration of use, and the part of the body the device interacts with.
? - Mdr Cert")
The classification rules are outlined in Annex VIII of MDR 2017/745, and further guidance is provided in MDCG 2021-24: Guidance on Classification of Medical Devices.
- Class I (Low risk) – Non-invasive and short-term use devices (e.g., bandages).
- Class IIa (Medium risk) – Devices intended for short-term use, often invasive but with lower associated risk. (e.g., hearing aids).
- Class IIb (Higher risk) – Devices intended for long-term use or active devices delivering energy to the body. (e.g., ventilators, infusion pumps, implants).
- Class III (High risk) – Devices that are life-sustaining, implanted long-term, or involve human tissue/cells. Life-sustaining devices (e.g., Pacemakers, heart valves).
Who Must Comply with MDR?
- Manufacturers – Must ensure compliance before CE marking.
- Authorized Representatives – Required for non-EU manufacturers.
- Importers & Distributors – Must verify device compliance.
Core Requirements of MDR
- Device Classification and Conformity Assessment – Determines the level of scrutiny (self-certification vs. notified body review).
- Technical Documentation Requirements- Includes MDR technical file and design dossier with clinical data.
- Clinical Evaluation and Investigation – Mandatory for most devices, with ongoing updates. (Keywords: clinical evaluation MDR)
- Post-Market Surveillance (PMS) and Vigilance – Requires PMS plans, PSURs, and incident reporting. (Keywords: post-market surveillance MDR, vigilance reporting)
Steps to Achieve MDR Compliance
Achieving compliance with MDR 2017/745 is a comprehensive process that requires careful planning, technical preparation, and collaboration across teams. Below are the key steps medical device manufacturers must follow, with a focus on strategic execution and regulatory alignment.
Conduct a Gap Analysis
Identify gaps between your current processes (based on MDD or other standards) and the new MDR requirements.
- Review your existing technical documentation, risk management files, and clinical evaluation reports.
- Compare each element against MDR requirements, particularly those found in Annex I (General Safety and Performance Requirements) and Annex II (Technical Documentation).
- Include an assessment of your current Quality Management System (QMS) and Post-Market Surveillance (PMS) plans.
Update Your Quality Management System (QMS)
Ensure your QMS aligns with ISO 13485:2016 and supports the broader requirements of MDR.
- Integrate MDR-specific processes into your QMS, such as Unique Device Identification (UDI), Eudamed registration, vigilance reporting, and clinical evaluation planning.
- Ensure proper documentation of corrective and preventive actions (CAPAs), supplier control, and product lifecycle traceability.
Prepare MDR-Compliant Technical Documentation
Build a comprehensive and structured technical file (Annex II + III) that meets MDR standards.
Key elements include:
- Device description and specifications
- Design and manufacturing information
- General Safety and Performance Requirements (GSPRs) checklist
- Risk management and usability engineering reports
- Clinical evaluation report (CER)
- Post-market surveillance (PMS) and Post-Market Clinical Follow-Up (PMCF) plans
Engage a Notified Body (For Class IIa, IIb, III Devices)
Obtain conformity assessment and CE marking through a designated Notified Body (NB).
- Choose a Notified Body designated under MDR for your device’s scope and classification.
- Prepare for audits and reviews of your technical documentation and QMS.
- Be ready to provide clinical evidence, PMS plans, and labeling samples.
Tip: Start early—NB capacity is limited and the review process can take several months.
Timeline and Deadlines
The EU Medical Device Regulation (MDR 2017/745) officially replaced the Medical Device Directive (MDD) in May 2021. However, recognizing the significant compliance burden for manufacturers, the European Commission introduced extended transition periods for legacy devices (devices certified under the MDD or AIMDD before MDR’s full application).
This timeline is crucial for ensuring continued market access and avoiding disruptions in supply. Below is a breakdown of the key MDR transition deadlines every manufacturer should know.
May 26, 2021 – Full MDR Enforcement
- The MDR became fully applicable and legally binding across the European Union.
- From this date, all new medical devices placed on the market must comply with MDR requirements.
- Notified Bodies were only allowed to issue CE certificates under MDR, not MDD.
2024–2028 – Extended Transition Periods for Legacy Devices
In March 2023, the European Parliament adopted an amendment (Regulation (EU) 2023/607) that extended MDR transition deadlines for legacy devices, aiming to prevent shortages and maintain patient access to critical medical technologies.
New MDR Transition Deadlines (Depending on Device Class):
| Device Classification | Extended Deadline |
| Class III and IIb implants | 31 December 2027 |
| Class IIb (non-implant), IIa, I (sterile/measuring) | 31 December 2028 |
Conditions for Extension:
- The device must continue to comply with MDD or AIMDD requirements.
- No significant changes to the design or intended purpose.
- A transition plan must be in place, and the manufacturer must have applied for MDR conformity assessment before the deadline.
Top 5 MDR Implementation Challenges
Implementing MDR 2017/745 poses significant hurdles for medical device manufacturers. The regulation’s stricter requirements and broader scope have made compliance more complex, especially for companies transitioning from MDD. Below are the top 5 challenges faced during MDR implementation, along with targeted keywords to enhance your content’s search visibility.
Lack of Notified Body Capacity
- One of the most critical issues is the limited number of MDR-designated Notified Bodies.
- This has led to significant certification delays, especially for higher-risk Class IIb and III devices.
- Manufacturers struggle to secure audit slots, jeopardizing product continuity in the EU market.
Clinical Data Gaps
- MDR demands robust clinical evidence to demonstrate device safety and performance.
- Legacy devices often lack sufficient clinical data, requiring new clinical investigations or literature reviews.
- This challenge is particularly acute for Class III and implantable devices.
Supply Chain Complexity
- MDR expands compliance responsibilities to economic operators, including importers, distributors, and authorized representatives.
- Ensuring full supply chain alignment with MDR requirements (such as UDI, traceability, and labeling updates) is both time-consuming and resource-intensive.
- Inadequate documentation or communication across partners can result in compliance failures.
Post-Market Surveillance Burden
- MDR enforces proactive and continuous PMS activities, even for lower-risk devices.
- Manufacturers must create and maintain PMS plans, PMCF (Post-Market Clinical Follow-up) reports, and Periodic Safety Update Reports (PSURs).
- Smaller companies may lack the infrastructure or personnel to manage this volume of ongoing data collection and analysis.
Technical Documentation Overhaul
- MDR requires significantly more detailed and structured technical documentation than MDD.
- Many companies underestimate the time and effort needed to rebuild technical files in accordance with Annex II and III.
- Errors or omissions can result in audit findings and rejected submissions.
Benefits of MDR for Patient Safety and Industry
The EU Medical Device Regulation (MDR 2017/745) was introduced not just to increase regulatory oversight but to significantly enhance patient safety and bring greater transparency, accountability, and innovation to the medical device industry. Below are the two core benefits:
How MDR Improves Device Safety
One of the central goals of MDR is to protect patients by improving the safety and performance of medical devices throughout their lifecycle. Here’s how:
Stricter Clinical Evaluation Requirements
Under MDR, manufacturers must provide robust clinical evidence to prove that their devices are safe and perform as intended. This includes:
- Continuous clinical evaluation and post-market clinical follow-up (PMCF).
- Clear alignment with Annex XIV and MDCG guidance (e.g., MDCG 2020-13 for legacy devices).
- Use of real-world evidence and scientific literature to support clinical claims.
Result: Fewer unsafe or under-tested devices reach the market.
Reinforced Risk Management
MDR places risk management at the core of the design and development process:
- Manufacturers must comply with ISO 14971 and integrate risk assessments into the technical documentation.
- Lifecycle-based risk monitoring ensures hazards are mitigated proactively, not reactively.
Result: Better protection against device malfunctions and adverse events.
Improved Transparency and Traceability
With systems like UDI (Unique Device Identification) and EUDAMED, regulators, healthcare professionals, and patients can trace devices more effectively:
- Product recalls are faster and more efficient.
- Counterfeit products are easier to detect.
Market Advantages of Being MDR-Compliant
While MDR compliance requires significant effort, it also delivers strategic benefits for manufacturers. Companies that achieve compliance early gain a competitive edge in both the EU and international markets.
Access to the EU Market
The EU represents one of the world’s largest and most lucrative medical device markets. Without MDR certification, manufacturers cannot place devices on the EU market after the applicable transition deadlines.
Result: MDR compliance is a must for market access.
Competitive Differentiation
MDR certification signals to customers, investors, and stakeholders that your device meets high standards of quality, safety, and performance. This enhances:
- Brand reputation
- Market trust
- Sales opportunities, especially with hospitals and procurement agencies that favor MDR-compliant suppliers.
Readiness for Global Expansion
Since MDR is considered one of the most rigorous regulatory frameworks, achieving compliance also:
- Prepares manufacturers for other markets like Australia, Canada, and Japan.
- Strengthens your case for FDA approval by aligning internal processes and documentation.
MDR compliance is not just a legal obligation, but a strategic investment that enhances product reliability and competitive advantage. By leveraging official guidance documents and expert consultancy services throughout the process, you can ensure a successful transition.
Remember: Regulatory requirements are constantly evolving. Stay updated on changes to guarantee long-term compliance.

? - Mdr Cert")
 - Mdr Cert")
{kind=link}
{kind=link}