 - Mdr Cert")
What Is IVDR (In Vitro Diagnostic Regulation)?
The In Vitro Diagnostic Regulation (IVDR), officially known as EU In Vitro Diagnostic Regulation (IVDR 2017/746), is the European Union’s updated regulatory framework governing in vitro diagnostic (IVD) medical devices. Replacing the previous In Vitro Diagnostic Directive (IVDD 98/79/EC), the IVDR introduces stricter requirements to enhance patient safety, improve clinical evidence, and strengthen post-market surveillance.
Key aspects of IVDR include:
- Risk-based classification system (Class A to D)
- Stricter clinical evidence requirements
- Enhanced post-market monitoring
- Unique Device Identification (UDI) system for traceability
- More rigorous notified body oversight
Why IVDR Matters for In Vitro Diagnostic Device Manufacturers
The IVDR significantly impacts manufacturers by:
- Increasing compliance requirements – More stringent documentation and performance studies.
- Improving diagnostic reliability – Stronger clinical evidence reduces false results.
- Enhancing transparency – Better labeling and technical documentation.
- Ensuring market access – Non-compliance means losing EU market eligibility.
Manufacturers must adapt to these changes to remain competitive and legally compliant.
History of In Vitro Diagnostic Medical Device Legislation in the EU
The EU’s IVD regulations have evolved from:
- In Vitro Diagnostic Directive (IVDD, 1998)
- In Vitro Diagnostic Regulation (IVDR, 2017/746)
The IVDR (2017/746), fully applicable since May 2022, was introduced to address gaps in the IVDD, particularly concerning high-risk IVDs and performance evaluations.
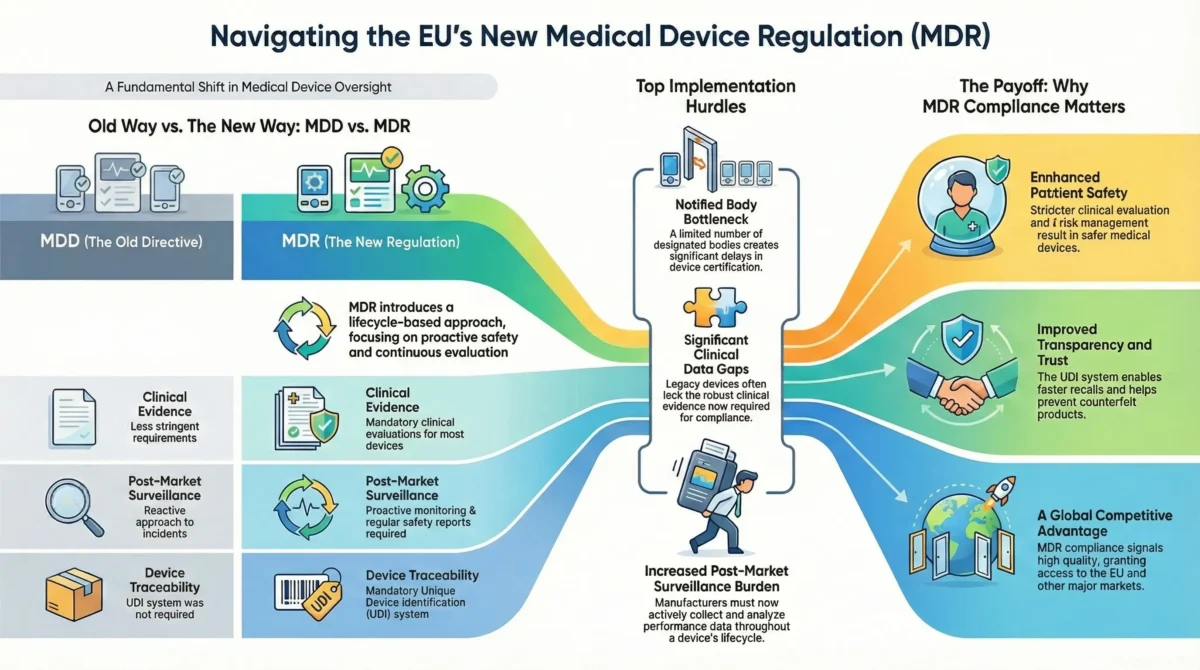
Key Differences Between IVDD and IVDR
| Aspect | IVDD (98/79/EC) | IVDR (2017/746) |
| Scope | Limited software regulation | Explicit inclusion of software as an IVD |
| Clinical Evidence | Less stringent requirements | Mandatory performance studies for most devices |
| Post-Market Surveillance (PMS) | Reactive approach | Proactive PMS & Post-Market Performance Follow-up (PMPF) |
| UDI System | Not required | Mandatory for traceability |
| Notified Body Oversight | Less rigorous | Stricter assessments for most classes |
Which Products Fall Under IVDR?
IVDR classifies devices into four risk classes:
- Class A (Low risk) – General lab equipment (e.g., buffers).
- Class B (Medium risk) – Pregnancy tests, cholesterol tests.
- Class C (High risk) – Cancer diagnostics, genetic tests.
- Class D (Highest risk) – HIV, hepatitis, blood grouping tests.
Who Must Comply with MDR?
- Manufacturers – Must ensure compliance before CE marking.
- Authorized Representatives – Required for non-EU manufacturers.
- Importers & Distributors – Must verify device compliance.
Core Requirements of MDR
- Device Classification and Conformity Assessment
Determines the level of scrutiny (self-certification vs. notified body review).
- Technical Documentation Requirements
Includes IVDR technical file and performance evaluation reports.
- Clinical Evaluation and Investigation
Mandatory for most devices, with ongoing updates.
- Post-Market Surveillance (PMS) and Vigilance
Requires PMS plans, PMPF, and incident reporting.
Steps to Achieve IVDR Compliance
- Gap Analysis – Compare existing processes vs. IVDR.
- Update QMS – Align with ISO 13485:2016.
- Prepare Technical Documentation – Including performance data.
- Engage a Notified Body – For Class B-D devices.
Timeline and Deadlines
- May 2022 – Full IVDR enforcement.
- 2025-2027 – Extended deadlines for legacy devices.
Update Your Quality Management System (QMS)
Ensure your QMS aligns with ISO 13485:2016 and supports the broader requirements of IVDR.
- Integrate IVDR-specific processes into your QMS, such as Unique Device Identification (UDI), Eudamed registration, vigilance reporting, and clinical evaluation planning.
- Ensure proper documentation of corrective and preventive actions (CAPAs), supplier control, and product lifecycle traceability.
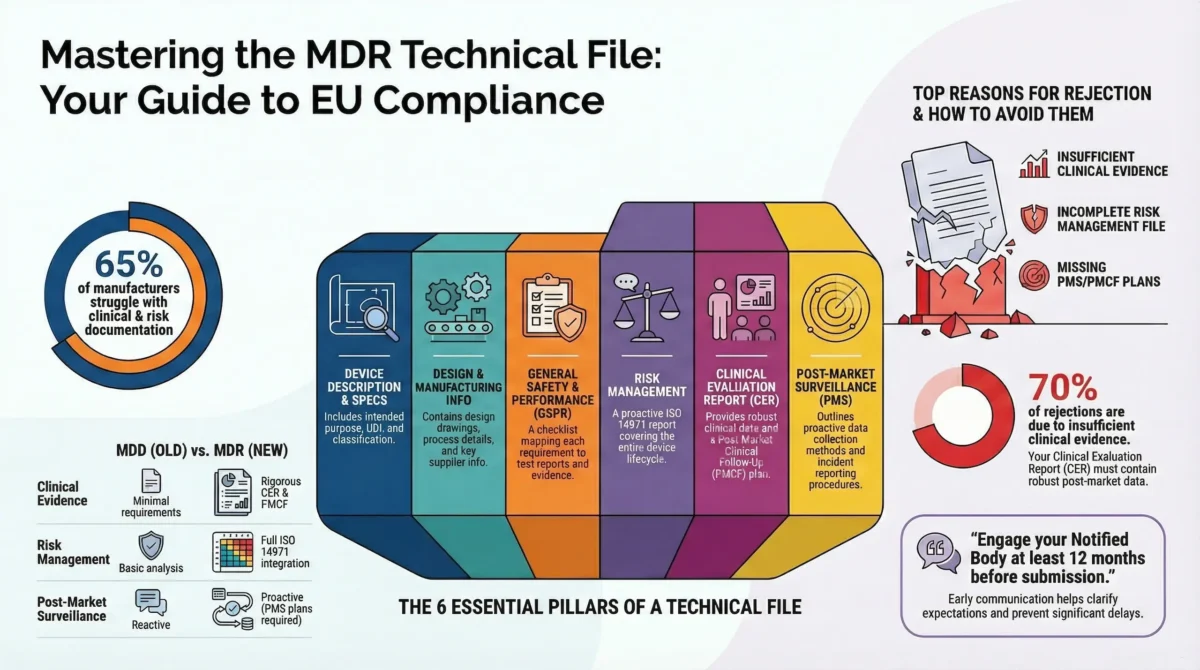
Prepare MDR-Compliant Technical Documentation
Build a comprehensive and structured technical file (Annex II + III) that meets MDRstandards.
Key elements include:
- Device description and specifications
- Design and manufacturing information
- General Safety and Performance Requirements (GSPRs) checklist
- Risk management and usability engineering reports
- Performance Evaluation Report (PER)
- Post-Market Surveillance (PMS) & Vigilance
Engage a Notified Body (For Class B, C, D IVD Devices)
Obtain conformity assessment and CE marking through a designated Notified Body (NB).
- Select an IVDR-Designated Notified Body
- Prepare for IVDR Audits & Documentation Review
- Expect Rigorous QMS Assessment.
Tip: Start early—NB capacity is limited and the review process can take several months.
Timeline and Deadlines
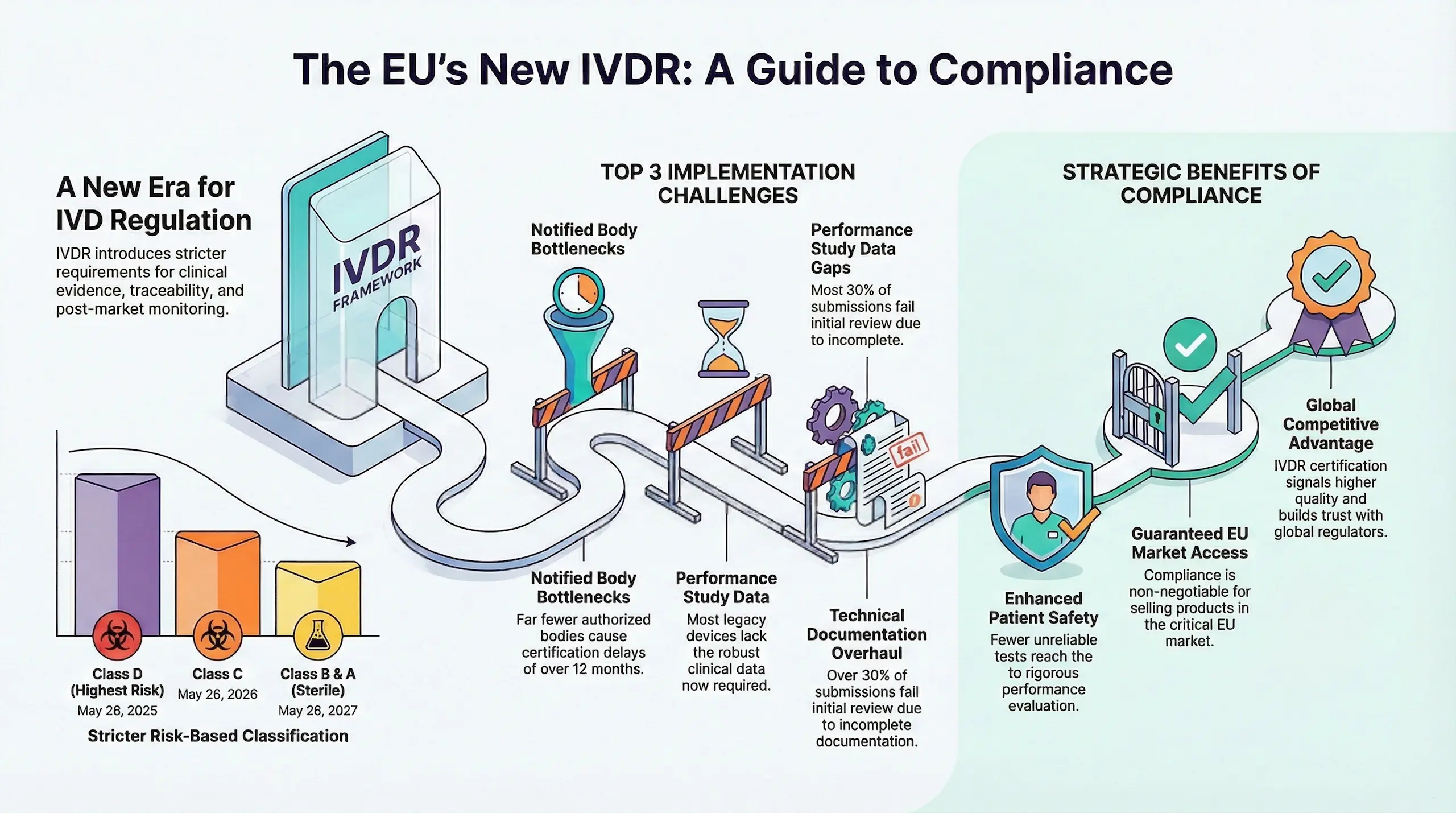
The In Vitro Diagnostic Regulation (IVDR 2017/746) fully replaced the IVD Directive (IVDD 98/79/EC) in May 2022. However, due to industry challenges, the EU introduced extended transition periods for legacy IVD devices.
May 26, 2022 – Full IVDR Enforcement
- All new IVDs entering the EU market must comply with IVDR
- Notified Bodies can only issue IVDR certificates (no new IVDD certifications)
2025–2027 – Extended Transition Periods
To prevent IVD shortages, Regulation (EU) 2022/112 extended deadlines for legacy devices:
New IVDR Transition Deadlines (Depending on Device Class):
| Device Classification | Extended Deadline |
| Class D (Highest Risk) | May 26, 2025 |
| Class C | May 26, 2026 |
| Class B & A (Sterile) | May 26, 2027 |
Conditions for Extension:
- Device remains IVDD-compliant (no significant changes)
- Manufacturer submits IVDR application before the deadline
- No safety concerns requiring immediate withdrawal
Top 5 MDR Implementation Challenges
Implementing IVDR 2017/746 presents significant hurdles for in vitro diagnostic (IVD) manufacturers. The regulation’s stricter clinical evidence requirements and expanded scope have made compliance more complex, especially for companies transitioning from IVDD. Below are the top 5 challenges faced during IVDR implementation, along with targeted keywords to enhance search visibility.
Notified Body Bottlenecks
- Limited IVDR-designated Notified Bodies – Far fewer NBs are authorized under IVDR compared to IVDD.
- Long certification delays – High-risk Class C & D devices face 12+ month review timelines.
- Risk of market exit – Companies that delay NB engagement may miss deadlines.
Solution: Apply early and choose an NB with relevant IVDR scope.
Performance Study Data Gaps
- Stricter performance evaluation – IVDR demands analytical + clinical performance data for most devices.
- Legacy IVDs lack sufficient evidence – Many IVDD-certified tests require new studies or literature reviews.
- Costly & time-consuming – Especially for Class D (e.g., HIV/hepatitis tests).
Solution: Conduct a gap analysis and prioritize high-risk devices first.
Supply Chain Compliance
- Expanded economic operator roles – Importers, distributors, and ARs must verify compliance.
- UDI & labeling updates – Full traceability is mandatory under IVDR.
- Contractual agreements needed – All partners must align on IVDR responsibilities.
Solution: Audit suppliers early and update contracts.
Post-Market Surveillance Burden
- Proactive PMS required – Even for Class A & B devices.
- Post-Market Performance Follow-Up (PMPF) – Mandatory for Class C & D.
- Periodic Safety Update Reports (PSURs) – Resource-intensive for small manufacturers.
Solution: Automate data collection where possible.
Technical Documentation Overhaul
- Annex II & III demand more detail – Performance evaluations, risk management, and manufacturing data must be exhaustive.
- Common pitfalls – Incomplete performance studies or outdated risk reports.
- Notified Body rejections – 30%+ of submissions fail initial review due to documentation gaps.
Solution: Use IVDR checklist templates and expert reviews.
Why These Challenges Matter
Non-compliance = EU market loss
Certification delays hurt revenue
Need IVDR support? MDRCert specializes in Notified Body coordination, performance evaluations, and documentation prep. Contact us for a compliance roadmap.
Benefits of MDR for Patient Safety and Industry
The EU In Vitro Diagnostic Regulation (IVDR 2017/746) was introduced not just to increase regulatory oversight but to significantly enhance diagnostic accuracy, patient safety, and industry transparency. Below are the two core benefits:
How IVDR Improves Diagnostic Safety
One of the central goals of IVDR is to protect patients by ensuring reliable and accurate diagnostic results throughout a device’s lifecycle. Here’s how:
Stricter Performance Evaluation Requirements
Under IVDR, manufacturers must provide robust clinical and analytical evidence to prove their IVDs perform as intended. This includes:
- Performance Evaluation Reports (PER) with scientific validity, analytical, and clinical performance data.
- Post-Market Performance Follow-Up (PMPF) for continuous monitoring.
- Alignment with MDCG guidance (e.g., MDCG 2022-2 on performance studies).
Result: Fewer unreliable or inaccurate tests reach the market.
Reinforced Risk Management
IVDR integrates risk management into every stage of device development:
- Compliance with ISO 14971 for systematic risk analysis.
- Lifecycle-based risk monitoring (not just pre-market).
Result: Reduced false positives/negatives and better patient outcomes.
Improved Transparency & Traceability
With systems like UDI (Unique Device Identification) and EUDAMED, stakeholders can:
- Track IVDs more efficiently.
- Detect counterfeit or unsafe tests faster.
Market Advantages of Being IVDR-Compliant
While IVDR compliance requires effort, it delivers strategic benefits for manufacturers. Companies achieving compliance early gain a competitive edge in global markets.
Guaranteed EU Market Access
The EU is a critical IVD market—without IVDR certification, manufacturers cannot sell tests after transition deadlines.
Result: Compliance is non-negotiable for continued market presence.
Competitive Differentiation
IVDR certification signals:
- Higher quality standards than IVDD.
- Greater trust from labs, hospitals, and regulators.
- Stronger brand reputation in a crowded market.
Global Expansion Readiness
Since IVDR is one of the strictest IVD regulations, compliance also:
- Prepares manufacturers for other markets (e.g., FDA, Health Canada).
- Streamlines future certifications by aligning processes early.
IVDR compliance is not just a regulatory requirement – it’s a strategic investment that strengthens diagnostic reliability and delivers long-term market advantages. By utilizing official MDCG guidance and expert consultancy support throughout your transition, you can ensure full compliance while optimizing your business strategy.
Remember: The IVDR landscape continues to evolve with new guidance and amendments. Proactive monitoring of regulatory updates is essential for maintaining continuous compliance and market access.

 - Mdr Cert")
? - Mdr Cert")
{kind=link}
{kind=link}