? - Mdr Cert")
? - Mdr Cert")
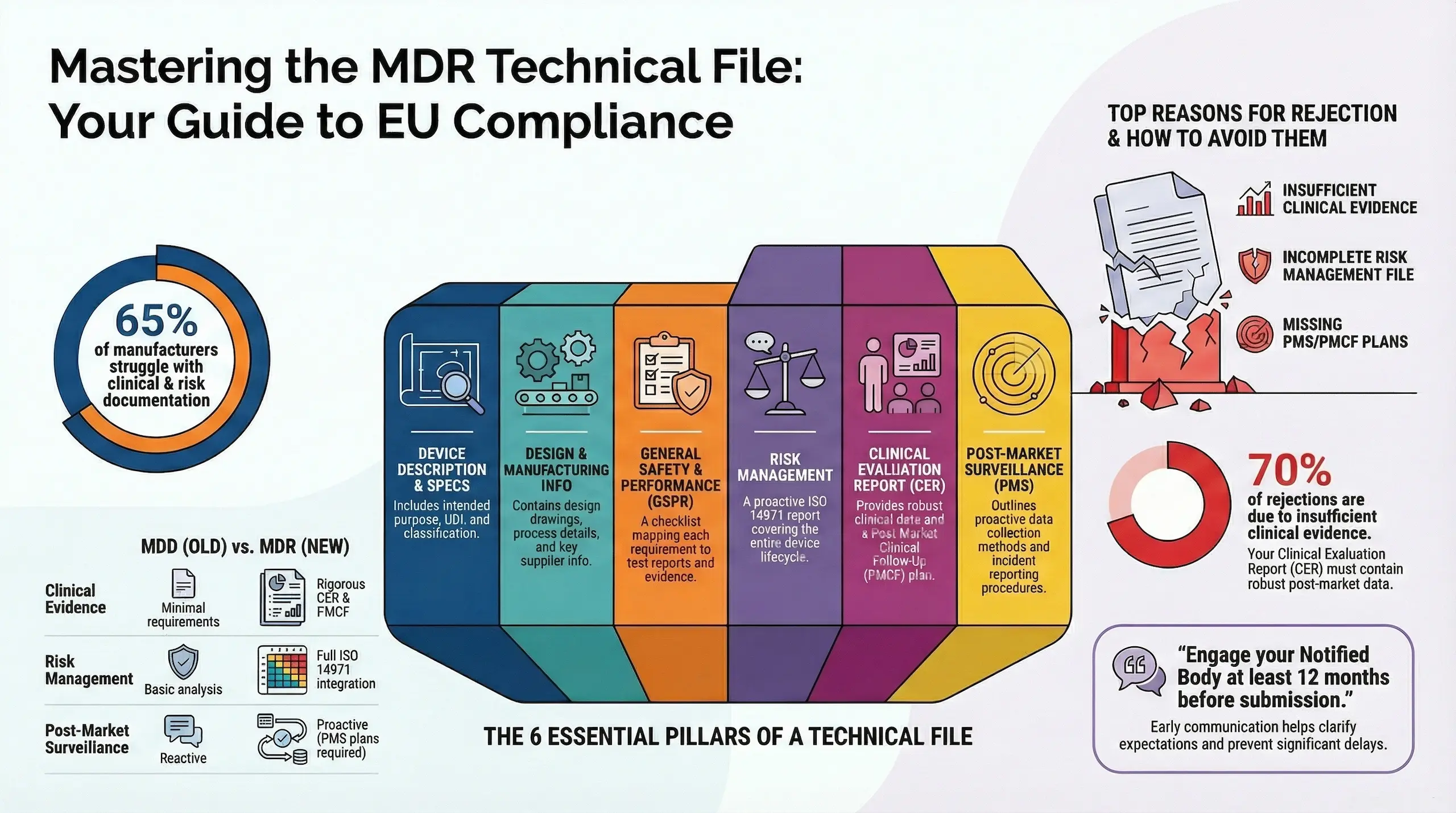
How to Structure an MDR-Compliant Technical File (Annex II & III)
Creating an MDR-compliant technical file is one of the most critical requirements under the EU Medical Device Regulation (2017/745/EU). It’s not just about satisfying regulatory authorities—it’s about proving your device is safe and effective for patients. Whether you’re a manufacturer or regulatory affairs professional, understanding how to properly assemble the technical documentation is key to getting your product approved for the EU market.
This guide provides a detailed, actionable breakdown of how to structure your technical file according to Annex II and Annex III of MDR. From essential documentation to post-market surveillance strategies, we cover everything you need to ensure compliance and avoid costly delays.
The EU Medical Device Regulation (MDR 2017/745) requires manufacturers to submit a detailed technical file (Annex II & III) for CE marking. Unlike the MDD, the MDR demands stricter clinical evidence, risk management, and post-market surveillance—making compliance more complex.
Why is this important?
- Without a properly structured technical file, your device won’t receive CE certification.
- Notified Bodies (NBs) rigorously review documentation—30% of submissions fail due to incomplete files.
- A well-prepared technical file reduces audit delays and ensures faster market access.
Did You Know?
According to a 2023 survey, 65% of manufacturers struggled most with clinical evaluations and risk management documentation under MDR.
What is an MDR Technical File?
A technical file under MDR (2017/745) is a comprehensive collection of documents proving that a medical device meets all applicable safety and performance requirements. It is essential for CE marking and must be made available to the Notified Body and Competent Authorities upon request.
Key Components of an MDR-Compliant Technical File
Device Description & Specifications
This section must include:
- Intended purpose & indications for use
- Device classification (Class I, IIa, IIb, III)
- UDI (Unique Device Identification) details
- Reference to previous generations (if applicable)
Design & Manufacturing Information
- Design drawings & schematics
- Manufacturing process (GMP compliance)
- Sterilization & shelf-life validation
- Critical suppliers & subcontractors
General Safety & Performance Requirements (GSPR) Checklist
Mapped to Annex I of MDR:
- Annex I compliance (all applicable GSPRs)
- Justification for each requirement
- Reference to supporting test reports
Example:
| GSPR Requirement | Compliance Evidence |
| Chemical Safety | Biocompatibility test report (ISO 10993) |
Risk Management & Usability Engineering
- ISO 14971:2019 Risk Management Report
- Use-error analysis (Human Factors Engineering)
- Mitigation strategies
Expert Quote:
“Under MDR, risk management must be proactive, not reactive—manufacturers must document hazards throughout the device lifecycle.”
Clinical Evaluation Report (CER)
- Clinical data from equivalent devices
- Post-Market Clinical Follow-Up (PMCF) plan
- MDCG 2020-13 compliance for legacy devices
📌 Statistic: *70% of CER rejections are due to insufficient post-market data.*
Product Verification & Validation Data
- Bench testing results
- Biocompatibility data (ISO 10993)
- Software validation
- Clinical evaluation report (CER)
Post-Market Surveillance (PMS) & PMCF Plans
- PMS Plan (Proactive monitoring) – Data collection sources (complaints, vigilance reports, literature), Analysis methodology and frequency, Proactive and reactive surveillance
- Periodic Safety Update Reports (PSURs) – PMCF rationale and objectives, Evaluation methods (surveys, studies, registry data), PMCF Evaluation Report (PMCF ER)
- Incident reporting procedures
Common Mistakes & How to Avoid Them
| Mistake | How to Avoid |
| Missing UDI or classification | Always include Annex VIII rationale in section 1 |
| Incomplete GSPR checklist | Map each requirement with linked evidence |
| Lack of clinical evidence | Include robust CER & PMS data |
| Incomplete risk management files | Use ISO 14971 templates. |
| Lack of PMCF data | Start collecting real-world evidence early. |
| Poor document organization | Follow Annex II & III structure strictly. |
Notified Body Review Process
- Pre-submission meeting (Discuss scope & expectations)
- Documentation review (3-6 months for Class III devices)
- On-site audit (If required)
- CE certificate issuance
📌 Pro Tip: Engage your NB at least 12 months before submission.
Key Takeaways
- Start early—Notified Body delays are common.
- Annex II outlines what must be in your technical file.
- Annex III focuses on PMS and PMCF documentation.
- Proper documentation = faster CE marking and fewer audits.
- Use templates, checklists, and indexes to stay organized.
Conclusion & Next Steps
Creating an MDR-compliant technical file is complex but critical for EU market access. To simplify the process:
- Conduct a gap analysis of your current documentation.
- Use templates for CER, PMS, and risk management.
- Consult experts to avoid costly mistakes.
Need help? Contact MDRCert for end-to-end MDR technical file support.
FAQs on MDR Technical Documentation
What documents are mandatory in an MDR technical file?
The essential documents include:
- Device description & specifications
- Design & manufacturing information
- General Safety & Performance Requirements (GSPR) checklist
- Risk management report (ISO 14971)
- Clinical Evaluation Report (CER)
- Post-Market Surveillance (PMS) & PMCF plans
How does MDR technical documentation differ from MDD?
| Aspect | MDD | MDR |
| Clinical Evidence | Minimal requirements | Rigorous CER & PMCF |
| Risk Management | Basic analysis | Full ISO 14971 integration |
| Post-Market Surveillance | Reactive | Proactive (PMS plans required) |
| UDI | Not required | Mandatory |
Can I reuse my MDD technical file for MDR?
No—significant updates are needed, especially for:
- Clinical data (Must meet MDR’s stricter requirements)
- Risk management (Must align with ISO 14971:2019)
- PMS plans (Must include PMCF for most devices)
Tip: Ondurt a gap analysis to identify required changes.
How long should I retain MDR technical documentation?
- At least 10 years after the last device is placed on the market.
- 15 years for implantable devices.
Statistic: 40% of audit findings relate to incomplete document retention.
Who is responsible for maintaining the technical file?
- Manufacturers (Primary responsibility)
- Authorized Representatives (For non-EU companies)
- Notified Bodies (Review during audits)
What’s the most common reason for Notified Body rejections?
Top 3 reasons:
- Insufficient clinical evidence (70% of rejections)
- Incomplete risk management file
- Missing PMS/PMCF plans
Expert Tip:
*”Always cross-check your CER against MDCG 2020-13 for legacy devices.”*
Is a CER required for all device classes?
- Class I (non-sterile/non-measuring): Minimal clinical data.
- Class IIa/IIb/III: Full CER + PMCF required.
Are there templates for MDR technical documentation?
Yes! Official resources include:
- MDCG guidance documents (e.g., MDCG 2020-13)
- ISO/TR 20416 (Post-market surveillance template)
- Notified Body checklists (e.g., BSI, TÜV SÜD)

? - Mdr Cert")
? - Mdr Cert")
{kind=link}
{kind=link}